


Abstract:
Research at Kanazawa University, Theragen Etex Bio Institute and Seoul National University as reported in Nature Communications points towards pathways for the metastasis and malignant transitions that result from changes in the protein p53. The results suggest that the cooperative development of mutations in the proteins helps tumours spread and metastasis.
Understanding the genetic mutations and protein changes that take place in the progression of cancer is key to its treatment. Mutations in the gene TP53 and concomitant mutant p53 proteins in cancer cells have become notorious over the course of multiple studies of several different types of cancers. Yet so far, the mechanisms behind the role of these protein changes in propagating the disease has been poorly understood. Masanobu Oshima at Kanazawa University and his collaborators now report results that suggest just how these protein changes may lead to accelerated tumorigenesis and metastasis.
Previous studies of cancers in the lungs, intestines and pancreas, as well as colorectal cancer, had suggested that changes in p53 lead to invasive abilities and stem cell characteristics crucial for late-stage tumorigenesis. In addition to this “gain of function (GOF)” mutation, wild-type protein p53 is reduced in cancer cells through a “loss of heterozygosity” process, where instead of having two alleles for each gene corresponding to each parent, the gene has just one.
Oshima collaborated with researchers at Kanazawa University in Japan, Theragen Etex Bio Institute and Seoul National University in South Korea. They found that tumor cells that carry GOF mutant p53 and lost wild-type p53 are enriched in liver metastasis lesions, suggesting combination of p53 LOH and GOF mutations promotes metastasis (Fig. 1). In studies of single cells dissociated from each other, the researchers found that loss of heterozygosity was key to the cell’s survival and proliferation, which has implications for metastasis. In addition, the structures formed were more complex and glandular.
Further studies suggested that the combination of gain of function mutations and loss of heterozygosity provided some protection against cell death. When injected in live mouse models the combination also accelerated tumorigenesis as well. The tumours formed had a fibrotic microenvironment, a signature of a poor prognosis for colorectal cancer.
Fibrotic structures are also linked with inflammation pathways. The researchers analysed the mutant proteins with the ingenuity pathway analysis software package, which provides integrated analysis of gene expression and other cell traits. The analysis indicated growth factor/MAPK and inflammatory pathways were indeed upregulated in cells with gain of function mutations and loss of heterozygosity. What is more trametinib treatment, (which inhibits a protein in the MAPK pathway) suppressed the cloning efficiency of these mutant cells. Ingenuity pathway analysis also revealed that stem cell pathways were up regulated in cells with gain of function mutations and loss of heterozygosity, as well as cells where both Trp53 alleles were disrupted (Fig. 2). In their report, the researchers suggest their findings “will contribute to the future development of therapeutic strategies against CRC [colorectal cancer] metastasis.”
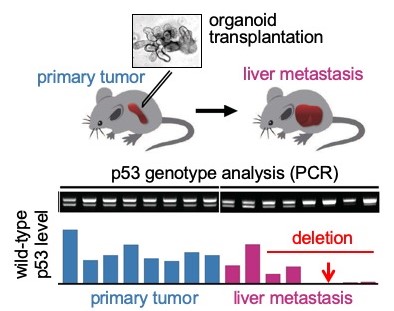
Figure 1
Tumor-derived organoids were transplanted to mouse spleen (top), and p53 status of the liver metastasized tumor cells were analyzed b PCR (middle). Notably, most metastasized tumor cells lost wild-type p53, suggesting that loss of p53 with GOF mutant p53 accelerates liver metastasis.
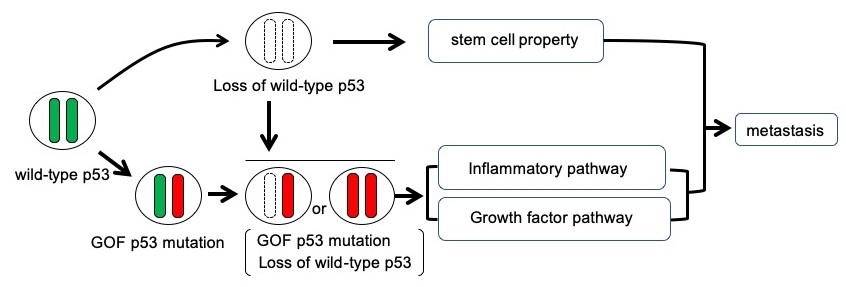
Figure 2
Schematic illustration of the p53 mutation and metastasis. Combination of loss of wild-type p53 and GOF p53 mutation promotes metastasis through acquisition of stem cell property and activation of inflammatory and growth factor pathways.
[Background]
Colorectal cancer
Colorectal cancer is one of the most fatal cancers world-wide. Not only are TP53 genetic mutations found in 55-60% of colorectal cancers but the percentage rises to 80% in metastasis-related cases of colorectal cancer, implying it has a role in the malignant progression of the cancer.
Tumorigenesis
In tumour initiation healthy cells become cancerous – their genetic make-up mutates and abnormal cell division processes take over. In healthy cells, proliferation and apoptosis (programmed cell death) balance. The imbalance between these processes is thought to be key to the onset and proliferation of cancer.
The researchers’ studies of the cells with respect to a process that causes intestinal epithelial cell death – anoikis – revealed that the combination of gain of function mutations and loss of heterozygosity provided cells with a level of protection. This could knock out of kilter the balance of production and destruction of cells – so delicately tuned in healthy cells.
Metastasis
Metastasis describes the spread of cancer to different parts of the body often through the lymph system or bloodstream. Tumours then develop at new sites from just a handful of cells.
The study of how cells that had gain of function mutations in the p53 protein fared once dissociated from each other has implications for metastasis. In fact the researchers found little difference between cells with or without loss of heterozygosity when they mechanically dissociated them. Here the dissociated cells remain as tumour glands rather than single cells. The real difference was observed when they used enzymes to dissociate them into single cells, and the single cells were subcloned (a technique for moving DNA into daughter cells). The subcloned population lost their wild-type Trp53, even when subcloned from cells with heterozygous Trp53 gain of function mutations.
Article
Loss of wild-type p53 promotes mutant p53-driven metastasis through acquisition of survival and tumor-initiating properties
Journal: Nature Communications
Authors: Mizuho Nakayama, Chang Pyo Hong, Hiroko Oshima, Eri Sakai, Seong-Jin Kim, Masanobu Oshima
DOI: 10.1038/s41467-020-16245-1
Funder
This work was supported by AMED (19ck0106259h0003) , Japan; and Grants-in-Aid for Scientific Research (18H04030, 17K07162, 16H06279) , MEXT, Japan.